Objetivo: provar con HEKs un nuevo método de IP (Pierce Magnetic IP kit) y compararlo con el método usado hasta ahora (Agarosa)
Diseño experimento: se harán 3 condiciones por cada método (se harán los dos métodos en paralelo)
- 4 placas –> HEK transfectadas con CNTN1 y tratadas con suero CNTN1 +
- 4 placas –> HEK transfectadas con CNTN1 y tratadas con suero CNTN1 –
- 2 placas –> HEK no transfectadas y tratadas con suero CNTN1+
15.10.2018: Coating HEK293
- Se preparan 20 placas de 100 mm –> coating con Poly-D-lisina 1/40 en Borate Buffer (1h a 37ºC) *En una de las placas se ponen dos cubreobjetos para comprovar la transfección.
- Se siembran 3 millones de cels/placa
16.10.2018: Transfección CNTN1
- Se transfectan 16 placas
- Por cada placa –> (13’4 µg DNA + 450 µl Optimem) + (20 µl lipofectamina + 450 µl Optimem)
- [CNTN1] = 1’311 µg/µl
- 160 µl DNA + 7 ml Optimem
- 320 µl lipofectamina + 7 ml Optimem
- [CNTN1] = 1’311 µg/µl
- Se incuba por separado durante 5 minutos a Tambiente y después se mezcla el DNA+lipo y se incuba 30 minutos a Tambiente
- Poner 0’9 ml de la mezcla a cada placa
17.10.2018: IP
Protocolo Agarosa
- Incubar el cultivo celular con el suero del paciente a analizar 1h a 37ºC (en el incubador). La dilución se hace en el propio medio de cultivo:
- Para IgG: 1/100 –> 30 µl de suero en 3 ml de medio
- Para IgG: 1/100 –> 30 µl de suero en 3 ml de medio
*La dilución se hace en la misma placa de cultivo (sacar el medio hasta dejar 3 ml por placa, y poner el suero)
- Cuando queden 5 min, preparar la mezcla Buffer de lisis + inhibidores de proteasas.
- Lavar las placas 2 veces con PBS atemperado, vaciar y añadir 800 µl de buffer lisis + inhibidores de proteasas a cada placa. Dejar en agitación fuerte (fijadas con cinta) durante 1h a 4ºC.
- En este intervalo de tiempo, hacer el lavado de la proteína A+G (INVITROGEN Cat:15920-010. y cat:15918-014). Preparar la Proteína A+G:
- Resuspender las bolas con agitación manual (no vórtex ni inversión.
- En un eppendorf por cada suero a analizar, añadir con punta de pipeta cortada 50 µl de proteína G + 50 µl de proteína A + 800 µl de buffer lisis
- Lavar 2 veces con Buffer de lisis centrifugando a 1000 rpm 5min. a 4ºC. Repetir un último lavado adicional añadiendo inhibidores de proteasas en el último lavado (del BL+IP sobrante del paso 2).
- Pasar el scraper a cada una de las placas y recoger todo el volumen y restos celulares en un eppendorf por placa.
- Centrifugar 5 min a velocidad máxima a 4ºC.
- Pasar el sobrenadante de todos los eppendorfs a un tubo de 15ml. Añadir 500 µl de buffer lisis con las bolas de proteína A+G sin pipetear arriba y abajo.
- Dejar O/N en rotación a 4ºC tapando el tubo con Parafilm.
- Al dia siguiente:
- Centrifugar el tubo de 15ml que dejamos en rotación con la proteína A+G a 3000 rpm 5 min a 4ºC.
- Descartar el sobrenadante y pasar el pellet con punta cortada a un eppendorf.
- Lavar el pellet 2 veces con 1 mL de lisis buffer. Centrifugar a 3000 rpm 5min a 4ºC después de cada lavado.
- Mientras preparar mezcla Laemmli + B-mercapto: 950 µL Laemmli+ 50 µL de B-mercapto
- Descartar sobrenadante y añadir al pellet 40 µl de Laemmli + Mercapto. Homogeneizar dando suaves golpes al eppendorf. Dejar 5 min a Tºambiente
- Calentar a 100ºC 5min (en el baño seco). Al acabar, atemperar la muestra 2 min a temperatura ambiente.
- Realizar el gel de acrilamida
Protocolo Pierce Magnetic IP Kit
- Incubar el cultivo con el suero del paciente a analizar 1h a 37ºC (en el incubador). La dilución se hace en el propio medio de cultivo (en la placa)
- Para IgG: 1/100 –> 30 µl de suero en 3 ml de medio
*La dilución se hace en la misma placa de cultivo (sacar el medio hasta dejar 3 ml por placa, y poner el suero)
- Lavar placas 1 vez con PBS 1x
- Añadir 800 µl de IP Lysis/Wash buffer + inhibidores de proteasas a cada placa. Dejar en agitación fuerte (fijadas con cinta) durante 5 min a 4ºC.
- Transferir el lisado a un eppendorf de 2 ml (en cada eppendorf de 2 ml cabe el lisado de dos placas) y centrifugar 10 minutos a 13000 g
- Poner 25 µl de Pierce Protein A/G Magnetic Beads a cada eppendorf
- Añadir 175 µl de IP Lysis/Wash Buffer y hacer vórtex (suave)
- Poner el tubo en el soporte magnético. Eliminar el sobrenadante (sin retirar el eppendorf)
- Añadir 1 ml de IP Lysis/Wash Buffer y hacer vórtex (suave). Poner de nuevo en el soporte magnético y eliminar el sobrenadante.
- Añadir la mezcla antígeno-anticuerpo (primera parte) al eppendorf con las bolas magnéticas lavadas e incubar a temperatura ambiente 1 hora en agitación (rotación).
- Poner el eppendorf en el soporte magnético y eliminar el sobrenadante.
- Añadir 500 µl de IP Lysis/Wash buffer (con inhibidores de proteasas) y mezclar. Poner en el soporte magnético y eliminar el sobrenadante.
- Volver a repetir el lavado
- Añadir 500 µl de agua destilada ultrapura y mezclar. Poner en el soporte magnético y eliminar el sobrenadante.
- Añadir 100 µl de Lane Marker Sample Buffer (diluida 5x en agua destilada) al tubo y calentar a 96 – 100 ºC durante 10 minutos.
- Poner en el soporte magnético para que las bolas magnéticas se separen.
- Con el sobrenadante realizar la electroforesis en gel de acrilamida.
18.10.2018: Electroforesis gel acrilamida
- Preparar el gel de resolución al 10 % y gel de concentración al 4 %
- Carregar les mostres (40 µl de cada)
- Laemmli
- Marcador
- Transfectadas (suero CNTN1+) AGAROSA
- Transfectadas (suero CNTN1-) AGAROSA
- No transfectadas (suero CNTN1+) AGAROSA
- Laemmli
- Transfectadas (suero CNTN1+) MAGNETIC BEADS
- Transfectadas (suero CNTN1-) MAGNETIC BEADS
- No transfectadas (suero CNTN1+) MAGNETIC BEADS
- Laemmli
- Correr el gel a 20 mA
- Parar la electroforesis y lavar el gel con 2 veces con H2Od
- Fijar durante 15 minutos
- 50 % metanol (10 ml)
- 10 % ácido acético (2 ml)
- 40 % H2Od (8 ml)
- Lavar 3 veces con H2Od
- Teñir con Novex (de 3 a 12 horas a Tambiente en agitación)
- 4 ml metanol
- 1 ml Stainer B
- 4 ml Stainer A
- 11 ml H2Od
- Desteñir lavando con H2Od overnight a 4ºC (en agitación)
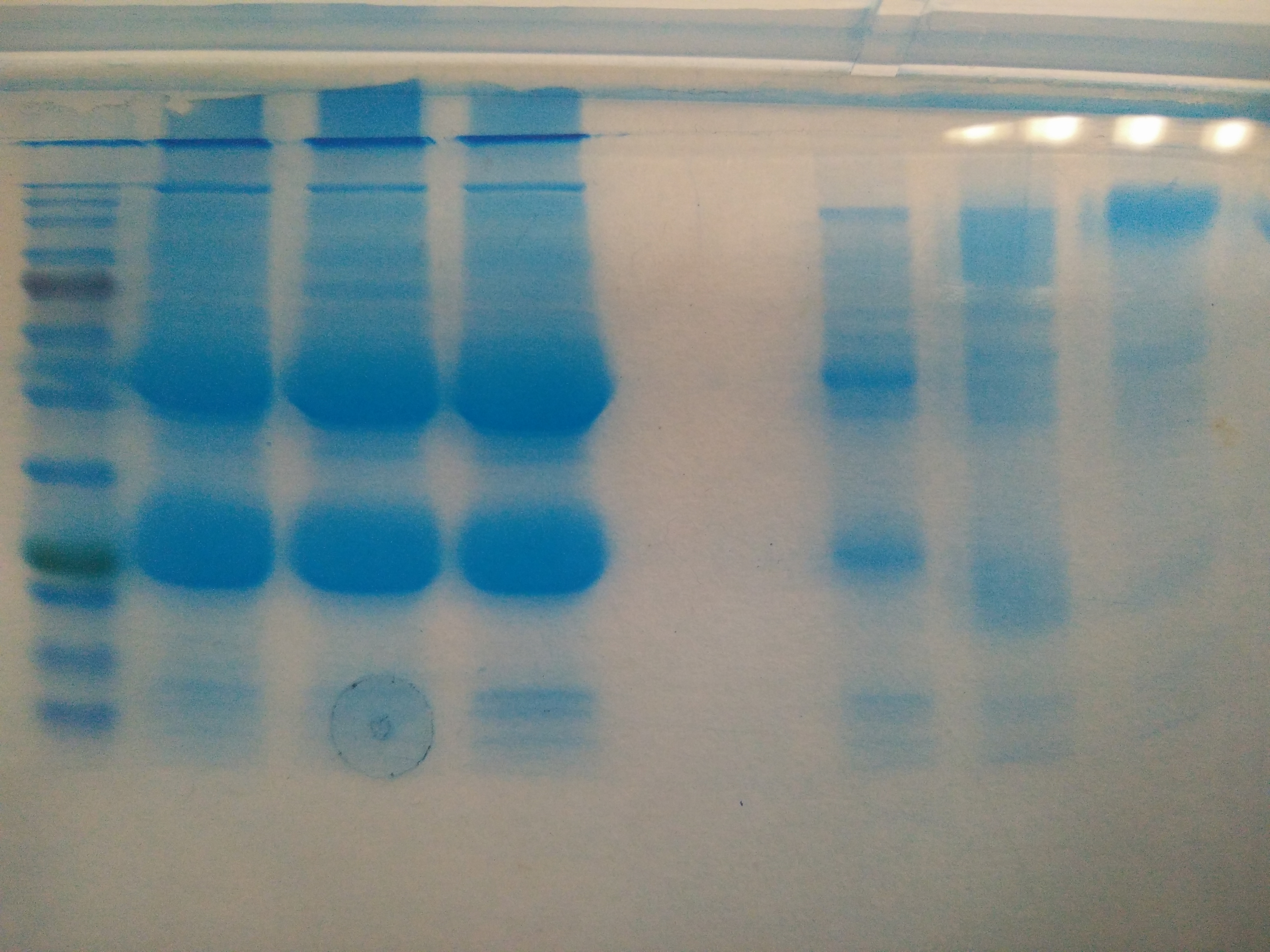
RESULTADO
A simple vista no se ven diferencias claras entre los pocillos con HEKs transfectadas tratadas con suero CNTN1+ y el resto de pocillos.
Entre los dos tipos de IP sí que hay diferencias –> en los pocillos de las meustras immunoprecipitadas con agarosa hay mucha más cantidad de proteína –> parece que la immunoprecipitación con magnetic beads sea más limpia.
Hacer western blot con las mismas muestras para ver si se observa la CNTN1.